
On this page:
- What is Usher syndrome?
- Who is affected by Usher syndrome?
- What causes Usher syndrome?
- What are the characteristics of the three types of Usher syndrome?
- How is Usher syndrome diagnosed?
- How is Usher syndrome treated?
- What research is being conducted on Usher syndrome?
- Where can I get more information about Usher syndrome?
What is Usher syndrome?
Usher syndrome is the most common condition that affects both hearing and vision; sometimes it also affects balance. The major symptoms of Usher syndrome are deafness or hearing loss and an eye disease called retinitis pigmentosa (RP) [re-tin-EYE-tis pig-men-TOE-sa].
Deafness or hearing loss in Usher syndrome is caused by abnormal development of hair cells (sound receptor cells) in the inner ear. Most children with Usher syndrome are born with moderate to profound hearing loss, depending on the type. Less commonly, hearing loss from Usher syndrome appears during adolescence or later. Usher syndrome can also cause severe balance problems due to abnormal development of the vestibular hair cells, sensory cells that detect gravity and head movement.
RP initially causes night-blindness and a loss of peripheral (side) vision through the progressive degeneration of cells in the retina. The retina is the light-sensitive tissue at the back of the eye and is crucial for vision. As RP progresses, the field of vision narrows until only central vision remains, a condition called tunnel vision. Cysts in the macula [MAC-u-la] (the central part of the retina) and cataracts (clouding of the lens) can sometimes cause an early decline in central vision in people with Usher syndrome.
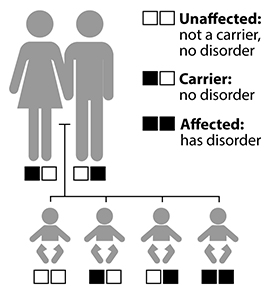
Chances of inheriting a recessive disorder
The hearing, balance, and vision of carriers with one mutant Usher gene is typically normal. Carriers are often unaware of their status.
Source: NIH/NIDCD
Who is affected by Usher syndrome?
Usher syndrome affects approximately 4 to 17 per 100,000 people,1, 2 and accounts for about 50 percent of all hereditary deaf-blindness cases.3 The condition is thought to account for 3 to 6 percent of all children who are deaf, and another 3 to 6 percent of children who are hard-of-hearing.4
What causes Usher syndrome?
Usher syndrome is inherited, which means that it is passed from parents to a child through genes. Each person inherits two copies of a gene, one from each parent. Sometimes genes are altered, or mutated. Mutated genes may cause cells to develop or act abnormally.
Usher syndrome is inherited as an autosomal recessive disorder. “Autosomal” means that men and women are equally likely to have the disorder and equally likely to pass it on to a child of either sex. “Recessive” means that the condition occurs only when a child inherits two copies of the same faulty gene, one from each parent. A person with one abnormal Usher gene does not have the disorder but is a carrier who has a 50 percent chance of passing on the abnormal gene to each child. When two carriers with the same mutated Usher syndrome gene have a child together, each birth has a:
- One-in-four chance of having a child who neither has Usher syndrome nor is a carrier.
- Two-in-four chance of having a child who is an unaffected carrier.
- One-in-four chance of having a child who has Usher syndrome.
What are the characteristics of the three types of Usher syndrome?
There are three types of Usher syndrome. In the United States, types 1 and 2 are the most common. Together, they account for up to 95 percent of Usher syndrome cases.5
Type 1: Children with type 1 Usher syndrome have profound hearing loss or deafness at birth and have severe balance problems. Many obtain little or no benefit from hearing aids but may be candidates for a cochlear implant—an electronic device that can provide a sense of sound to people with severe hearing loss or deafness. (For more information, read the NIDCD fact sheet Cochlear Implants.) Parents should consult with their child’s doctor and other hearing health professionals early to determine communication options for their child. Intervention should begin promptly, when the brain is most receptive to learning language, whether spoken or signed.
Balance problems associated with type 1 Usher syndrome delay sitting up without support. Walking rarely occurs prior to 18 months. Vision problems with type 1 Usher syndrome usually begin before age 10, starting with difficulty seeing at night and progressing to severe vision loss over several decades.
Type 2: Children with type 2 Usher syndrome are born with moderate to severe hearing loss but normal balance. Although the severity of hearing loss varies, most children with type 2 Usher syndrome can communicate orally and benefit from hearing aids. RP is usually diagnosed during late adolescence in people with type 2 Usher syndrome.
Type 3: Children with type 3 Usher syndrome have normal hearing at birth. Most have normal to near-normal balance, but some develop balance problems with age. Decline in hearing and vision varies. Children with type 3 Usher syndrome often develop hearing loss by adolescence, requiring hearing aids by mid-to-late adulthood. Night blindness also usually begins during adolescence. Blind spots appear by the late teens to early twenties. Legal blindness often occurs by midlife.
| Type 1 | Type 2 | Type 3 | |
|---|---|---|---|
| Hearing |
Profound hearing loss or deafness at birth. |
Moderate to severe hearing loss at birth. |
Progressive hearing loss in childhood or early teens. |
| Vision | Decreased night vision by age 10, progressing to severe vision loss by midlife. | Decreased night vision by adolescence, progressing to severe vision loss by midlife. |
Varies in severity and age of onset; night vision problems often begin in teens and progress to severe vision loss by midlife. |
| Balance (vestibular function) | Balance problems from birth. |
Normal balance. |
Normal to near-normal |
How is Usher syndrome diagnosed?
Diagnosis of Usher syndrome involves pertinent questions regarding the person’s medical history and testing of hearing, balance, and vision. Early diagnosis is important, as it improves treatment success. An eye care specialist can use dilating drops to examine the retina for signs of RP. Visual field testing measures peripheral vision. An electroretinogram [e-lec-tro-RET-in-o-gram] measures the electrical response of the eye’s light-sensitive cells in the retina. Optical coherence tomography may be helpful to assess for macular cystic changes. Videonystagmography [vi-de-o-nigh-stag-MAH-gra-fee] measures involuntary eye movements that could signify a balance problem. Audiology testing determines hearing sensitivity at a range of frequencies.
Genetic testing may help in diagnosing Usher syndrome. So far, researchers have found nine genes that cause Usher syndrome. Genetic testing is available for all of them:
- Type 1 Usher syndrome: MY07A, USH1C, CDH23, PCHD15, USH1G
- Type 2 Usher syndrome: USH2A, GPR98, DFNB31
- Type 3 Usher syndrome: CLRN1
Genetic testing for Usher syndrome may be available through clinical research studies. Search for “Usher syndrome” or “Usher genetic testing” at the NIH Clinical Trials website.
How is Usher syndrome treated?
Presently, there is no cure for Usher syndrome. Treatment involves managing hearing, vision, and balance problems. Early diagnosis helps tailor educational programs that consider the severity of hearing and vision loss and a child’s age and ability. Treatment and communication services may include hearing aids, assistive listening devices, cochlear implants, auditory (hearing) training, and/or learning American Sign Language. Independent-living training may include orientation and mobility training for balance problems, Braille instruction, and low-vision services.
Vitamin A may slow the progression of RP, according to results from a long-term clinical trial supported by the National Eye Institute and the Foundation Fighting Blindness.6 Based on the study, adults with a common form of RP may benefit from a daily supplement of 15,000 IU (international units) of the palmitate form of vitamin A. Patients should discuss this treatment option with their health care provider before proceeding. Because people with type 1 Usher syndrome did not take part in the study, high-dose vitamin A is not recommended for these patients.
General precautions for vitamin A supplementation:
- Do not substitute vitamin A palmitate with a beta-carotene supplement.
- Do not take vitamin A supplements greater than the recommended dose of 15,000 IU or modify your diet to select foods with high levels of vitamin A.
- Pregnant women should not take high-dose vitamin A supplements due to the increased risk of birth defects. Women considering pregnancy should stop taking high-dose vitamin A supplements for six months before trying to conceive.
What research is being conducted on Usher syndrome?
Researchers are trying to identify additional genes that cause Usher syndrome. Efforts will lead to improved genetic counseling and earlier diagnosis, and may eventually expand treatment options.
Scientists are also developing mouse models with characteristics similar to Usher syndrome. Research using mouse models will help determine the function of Usher genes and inform potential treatments.
Other areas of study include developing new methods for early identification of children with Usher syndrome, improving treatment strategies for children who use hearing aids or cochlear implants for hearing loss, and testing innovative intervention strategies to help slow or stop the progression of RP. Clinical researchers are also characterizing variability in balance among individuals with various types of Usher syndrome.
Visit the NIH Clinical Research Trials and You website to read about these and other clinical trials that are recruiting volunteers.
Where can I get more information about Usher syndrome?
The NIDCD maintains a directory of organizations that provide information on the normal and disordered processes of hearing, balance, taste, smell, voice, speech, and language.
For more information, contact us at:
NIDCD Information Clearinghouse
1 Communication Avenue
Bethesda, MD 20892-3456
Toll-free voice: (800) 241-1044
Toll-free TTY: (800) 241-1055
Email: nidcdinfo@nidcd.nih.gov
National Eye Institute
National Eye Institute Communications Office
31 Center Drive MSC 2510
Bethesda, MD 20892-2510
Phone: (301) 496-5248
Email: 2020@nei.nih.gov
https://nei.nih.gov
NIH Pub. No. 98-4291
December 2017
1 Boughman, J.A., et al. (1983). Usher syndrome: definition and estimate of prevalence from two high-risk populations. Journal of Chronic Diseases, 36(8), 595–603.
2 Kimberling, W., et al. (2010). Frequency of Usher syndrome in two pediatric populations: implications for genetic screening of deaf and hard of hearing children. Genetics in Medicine, 12(8), 512–516.
3 Ben-Rebeh, I., et al. (2016). Genetic analysis of Tunisian families with Usher syndrome type 1: toward improving early molecular diagnosis. Molecular Vision, 22, 827–835.
4 National Library of Medicine. Usher syndrome. Genetics Home Reference: Your Guide to Understanding Genetic Conditions.
5 Saihan, Z., Webster, A.R., Luxon, L., Bitner-Glindzicz, M. (2009). Update on Usher syndrome. Current Opinion in Neurology, 22(1), 19–27.
6 Berson, E.L. (1998). Treatment of retinitis pigmentosa with vitamin A. Digital Journal of Ophthalmology, 4(7).
*Note: PDF files require a viewer such as the free Adobe Reader![]() .
.