
En esta página:
- ¿Qué es el síndrome de Usher?
- ¿A quién afecta el síndrome de Usher?
- ¿Qué causa el síndrome de Usher?
- ¿Cuáles son las características de los tres tipos de síndrome de Usher?
- ¿Cómo se diagnostica el síndrome de Usher?
- ¿Cómo se trata el síndrome de Usher?
- ¿Qué investigaciones se están llevando a cabo sobre el síndrome de Usher?
- ¿Dónde puedo obtener más información sobre el síndrome de Usher?
¿Qué es el síndrome de Usher?
El síndrome de Usher es el trastorno más frecuente que afecta tanto la audición como la visión. A veces también afecta el equilibrio. Los principales síntomas del síndrome de Usher son la sordera o pérdida de audición y una enfermedad de los ojos llamada retinitis pigmentaria.
La sordera o pérdida de audición que se presenta con el síndrome de Usher es causada por el desarrollo anormal de las células ciliadas (las células receptoras del sonido) en el oído interno. La mayoría de los niños con síndrome de Usher nacen con pérdida de audición de moderada a profunda, según el tipo del síndrome que tengan. Con menos frecuencia, la pérdida de audición por el síndrome de Usher aparece durante la adolescencia o más adelante. El síndrome de Usher también puede causar problemas graves de equilibrio debido al desarrollo anormal de las células ciliadas vestibulares, es decir, las células sensoriales que detectan la gravedad y el movimiento de la cabeza.
La retinitis pigmentaria inicialmente causa ceguera nocturna y pérdida de la visión periférica (lateral) debido a la degeneración progresiva de las células en la retina. La retina es el tejido sensible a la luz en la parte posterior del ojo y es esencial para la visión. A medida que la retinitis pigmentaria progresa, el campo visual se hace más estrecho hasta que solo queda la visión central, lo que en inglés a veces llaman visión de túnel. A veces, los quistes en la mácula (la parte central de la retina) y las cataratas (opacidad del cristalino) pueden causar una disminución temprana de la visión central en las personas con síndrome de Usher.
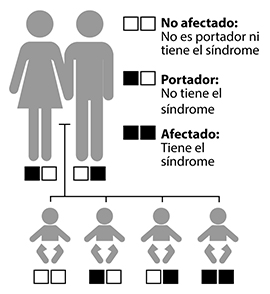
Probabilidades de heredar un trastorno recesivo
Por lo general, los portadores con un gen mutante de Usher tienen la audición, el equilibrio y la visión normales. A menudo, los portadores no saben que tienen un gen mutante.
Fuente: NIH/NIDCD
¿A quién afecta el síndrome de Usher?
El síndrome de Usher afecta a alrededor de 4 a 17 personas por cada 100,000,1, 2 y representa aproximadamente el 50 por ciento de todos los casos hereditarios de sordera con ceguera.3 Se cree que el síndrome representa entre el 3 y el 6 por ciento de todos los niños sordos y entre el 3 y el 6 por ciento de los niños con dificultad para oír.4
¿Qué causa el síndrome de Usher?
El síndrome de Usher es hereditario, lo que significa que se transmite de padres a hijos a través de los genes. Cada persona hereda dos copias de cada gen, una de su padre y una de su madre. A veces los genes se alteran o se mutan. Los genes mutados pueden hacer que las células se desarrollen o actúen de forma anormal.
El síndrome de Usher se hereda como un trastorno autosómico recesivo. "Autosómico" significa que tanto hombres como mujeres tienen la misma probabilidad de heredar el trastorno, así como la misma probabilidad de transmitirlo a un hijo de cualquier sexo. "Recesivo" significa que el síndrome ocurre solo cuando un niño hereda dos copias del mismo gen defectuoso, una copia del padre y una de la madre. Aunque una persona con un gen de Usher anormal no tiene el trastorno, sí es portador y tiene un 50 por ciento de probabilidad de transmitir el gen anormal a cada hijo. Cuando dos portadores con el mismo gen mutado del síndrome de Usher tienen un hijo juntos, cada nacimiento tiene las siguientes probabilidades:
- Una en cuatro de no tener síndrome de Usher ni ser portador.
- Dos en cuatro de ser portador, pero no tener el síndrome.
- Una en cuatro de tener el síndrome de Usher.
¿Cuáles son las características de los tres tipos de síndrome de Usher?
Hay tres tipos del síndrome de Usher. En los Estados Unidos, los tipos 1 y 2 son los más comunes. Juntos, representan hasta el 95 por ciento de los casos de síndrome de Usher.5
Tipo 1: los niños con síndrome de Usher tipo 1 tienen una profunda pérdida de audición o sordera al nacer y tienen graves problemas de equilibrio. Para muchos, los audífonos ofrecen poco o ningún beneficio, pero pueden ser candidatos para un implante coclear. Este es un aparato electrónico que puede dar una sensación de sonido a las personas con pérdida de audición grave o sordera. (Para más información, vea la hoja de información del NIDCD sobre Implantes cocleares). Los padres deben consultar con el médico de su hijo y otros profesionales de la salud auditiva lo más temprano posible para determinar las opciones de comunicación para su hijo. La intervención debe comenzar de inmediato, cuando el cerebro es más receptivo al aprendizaje del lenguaje, ya sea hablado o de señas.
Los problemas de equilibrio asociados con el síndrome de Usher tipo 1 retrasan que el niño pueda sentarse sin apoyo. Es raro que los niños puedan caminar antes de los 18 meses. Los problemas de visión con el síndrome de Usher tipo 1 usualmente se inician antes de los 10 años de edad, comenzando con dificultad para ver de noche y progresando a una pérdida de visión grave después de varias décadas.
Tipo 2: los niños con síndrome de Usher tipo 2 nacen con pérdida de audición de moderada a grave, pero con un equilibrio normal. Aunque la gravedad de la pérdida de audición varía, la mayoría de los niños con síndrome de Usher tipo 2 pueden comunicarse con lenguaje hablado y se benefician de los audífonos. En los niños con síndrome de Usher tipo 2, la retinitis pigmentaria se suele diagnosticar durante la adolescencia tardía.
Tipo 3: los niños con síndrome de Usher tipo 3 tienen audición normal al nacer. La mayoría tiene un equilibrio entre normal y casi normal, pero algunos comienzan a tener problemas de equilibrio con la edad. La disminución en la audición y la visión varía. Los niños con síndrome de Usher tipo 3 a menudo comienzan a perder la audición en la adolescencia y requieren audífonos a mediados o finales de la edad adulta. Por lo general, la ceguera nocturna también comienza en la adolescencia. Los puntos ciegos (o escotomas) aparecen a finales de la adolescencia hasta principios de los años veinte. La ceguera legal a menudo ocurre en la mediana edad.
| Tipo 1 | Tipo 2 | Tipo 3 | |
|---|---|---|---|
|
Audición |
Pérdida de audición profunda o sordera al nacer. |
Pérdida de audición de moderada a grave al nacer. |
Pérdida de audición progresiva en la niñez o adolescencia temprana. |
|
Visión |
Disminución de la visión nocturna a los 10 años, progresando a pérdida grave de la visión en la mediana |
Disminución de la visión nocturna en la adolescencia, progresando a pérdida grave de la visión en la mediana edad. |
Varía en gravedad y edad de inicio; los problemas de la visión nocturna a menudo comienzan en la adolescencia y progresan a la pérdida grave de la visión en la mediana edad. |
|
Equilibrio (función |
Problemas de equilibrio desde el nacimiento. |
Equilibrio normal. |
Equilibrio normal a casi normal en la infancia; posibilidad de problemas posteriores. |
¿Cómo se diagnostica el síndrome de Usher?
Para hacer un diagnóstico del síndrome de Usher, el médico debe hacer preguntas sobre la historia médica de la persona, así como pruebas de audición, equilibrio y visión. El diagnóstico temprano es importante, ya que mejora el éxito del tratamiento. Un especialista en el cuidado de los ojos puede usar gotas de dilatación para examinar la retina para buscar signos de retinitis pigmentaria. Las pruebas del campo visual miden la visión periférica. Un electrorretinograma mide la respuesta eléctrica de las células en la retina que son sensibles a la luz. La tomografía de coherencia óptica puede ser útil para evaluar los cambios en los quistes en la mácula. La videonistagmografía mide los movimientos involuntarios de los ojos que podrían significar un problema de equilibrio. Las pruebas de audiología determinan la sensibilidad auditiva en un rango de frecuencias.
Las pruebas genéticas pueden ayudar en el diagnóstico del síndrome de Usher. Hasta el momento, los investigadores han encontrado nueve genes que causan el síndrome de Usher. Hay pruebas genéticas para todos ellos:
- Síndrome de Usher tipo 1: MY07A, USH1C, CDH23, PCHD15, USH1G
- Síndrome de Usher tipo 2: USH2A, GPR98, DFNB31
- Síndrome de Usher tipo 3: CLRN1
Es posible que las pruebas genéticas para el síndrome de Usher estén disponibles a través de estudios de investigación clínica. Busque "Usher syndrome" o "Usher genetic testing" en el sitio web de NIH Clinical Trials.
¿Cómo se trata el síndrome de Usher?
Actualmente, no hay cura para el síndrome de Usher. El tratamiento implica el manejo de los problemas de audición, visión y equilibrio. El diagnóstico temprano ayuda a adaptar los programas educativos para que tomen en cuenta la gravedad de la pérdida de audición y visión y la edad y capacidad del niño. Los servicios de tratamiento y comunicación pueden incluir audífonos, aparatos de ayuda auditiva, implantes cocleares, capacitación auditiva y aprendizaje del lenguaje de señas americano. La capacitación para vivir de manera independiente puede incluir la capacitación para orientarse y movilizarse con problemas de equilibrio, instrucción en Braille y servicios de baja visión.
Según los resultados de un ensayo clínico a largo plazo respaldado por el Instituto Nacional del Ojo y la Fundación Fighting Blindness6, la vitamina A puede hacer que la retinitis pigmentaria avance más lentamente. Según el estudio, los adultos con una forma común de retinitis pigmentaria pueden beneficiarse de un suplemento diario de 15,000 UI (unidades internacionales) de la forma palmitato de vitamina A. Los pacientes deben hablar sobre esta opción de tratamiento con su médico u otro proveedor de atención médica antes de continuar. Debido a que las personas con síndrome de Usher tipo 1 no participaron en el estudio, no se recomienda una dosis alta de vitamina A para estos pacientes.
Precauciones generales sobre los suplementos de vitamina A:
- No sustituya el palmitato de vitamina A con un suplemento de betacaroteno.
- No tome suplementos de vitamina A en dosis más altas que la recomendada de 15,000 UI ni modifique su dieta para consumir alimentos con altos niveles de vitamina A.
- Las mujeres embarazadas no deben tomar suplementos de vitamina A en altas dosis debido a un mayor riesgo de defectos congénitos. Las mujeres que están considerando quedarse embarazadas deben dejar de tomar dosis altas de vitamina A seis meses antes de tratar de concebir.
¿Qué investigaciones se están llevando a cabo sobre el síndrome de Usher?
Los investigadores intentan identificar genes adicionales que causen el síndrome de Usher. Los esfuerzos darán lugar a una mejor asesoría genética y un diagnóstico más temprano, y con el tiempo podrán ampliar las opciones de tratamiento.
Los científicos también están desarrollando modelos en ratones con características similares al síndrome de Usher. Las investigaciones con modelos en ratones ayudarán a determinar qué función tienen los genes Usher e informarán sobre posibles tratamientos.
En otras áreas de estudio se intenta desarrollar nuevos métodos para la identificación temprana de niños con síndrome de Usher, mejorar las estrategias de tratamiento para niños que usan audífonos o implantes cocleares para la pérdida de audición y probar estrategias de intervención innovadoras para ayudar a frenar o detener el avance de la retinitis pigmentaria. Los investigadores clínicos también están caracterizando la variabilidad en el equilibrio en las personas con diferentes tipos de síndrome de Usher. Visite el sitio web de la investigación clínica de los NIH para leer sobre estos y otros ensayos clínicos que están recibiendo voluntarios.
¿Dónde puedo obtener más información sobre el síndrome de Usher?
El NIDCD mantiene un directorio de organizaciones que ofrecen información sobre los procesos normales y los trastornos de la audición, el equilibrio, el gusto, el olfato, la voz, el habla y el lenguaje. Actualmente, el directorio está disponible solamente en inglés.
Para más información, comuníquese con nosotros al:
Centro de Información del NIDCD
1 Communication Avenue
Bethesda, MD 20892-3456
Número de teléfono gratuito: (800) 241-1044
Número gratuito TTY: (800) 241-1055
Correo electrónico: nidcdinfo@nidcd.nih.gov
Instituto Nacional del Ojo
National Eye Institute Communications Office
31 Center Drive MSC 2510
Bethesda, MD 20892-2510
Número de teléfono: (301) 496-5248
Correo electrónico: 2020@nei.nih.gov
https://nei.nih.gov
Publicación de NIH núm. 98-4291 S
Diciembre de 2017
1 Boughman, J.A., et al. (1983). Usher syndrome: definition and estimate of prevalence from two high-risk populations. Journal of Chronic Diseases, 36(8), 595–603.
2 Kimberling, W., et al. (2010). Frequency of Usher syndrome in two pediatric populations: implications for genetic screening of deaf and hard of hearing children. Genetics in Medicine, 12(8), 512–516.
3 Ben-Rebeh, I., et al. (2016). Genetic analysis of Tunisian families with Usher syndrome type 1: toward improving early molecular diagnosis. Molecular Vision, 22, 827–835.
4 National Library of Medicine. Usher syndrome. Genetics Home Reference: Your Guide to Understanding Genetic Conditions.
5 Saihan, Z., Webster, A.R., Luxon, L., Bitner-Glindzicz, M. (2009). Update on Usher syndrome. Current Opinion in Neurology, 22(1), 19–27.
6 Berson, E.L. (1998). Treatment of retinitis pigmentosa with vitamin A. Digital Journal of Ophthalmology, 4(7).